
Épigénome et paléogénome
THIERRY GRANGE & EVA-MARIA GEIGL

Nous étudions l’évolution des génomes afin d’approfondir la compréhension de la relation entre les génotypes et les phénotypes. Les adaptations phénotypiques récentes sont idéales pour explorer l’évolution génomique sous-jacente tout en minimisant les effets confondants de la dérive génétique. Nous abordons ces aspects par l’analyse de génomes anciens provenant de fossiles, témoins directs de l’évolution, ajoutant ainsi une échelle de temps géologique ou historique aux études évolutives. Nous documentons les processus évolutifs survenus au cours des cent mille dernières années et repoussons les limites méthodologiques pour permettre l’étude d’échantillons plus anciens provenant de régions géographiques où le climat chaud est particulièrement préjudiciable à la préservation de l’ADN.
Mots-clés : évolution, génomes, ADN ancien, paléogénomique, peuplement, domestication
+33 (0)1 57 27 81 29 / +33 (0)1 57 27 81 32 Contact Thierry GRANGE / Contact Eva-Maria GEIGL @thierrygrange.bsky.social
Axes de recherche
Nous nous concentrons sur deux axes de recherche principaux : 1) la domestication des animaux, qui représente un modèle unique d’évolution accélérée de régions génomiques spécifiques sous l’impulsion humaine ; 2) l’évolution génomique des populations humaines au cours du peuplement de l’Europe au cours des 40 000 dernières années. Ces axes donnent lieu à cinq thèmes de recherche interdisciplinaires et interconnectés.
1.1 Étude paléogénomique de la dynamique passée des populations de bovins influencée par le climat et les humains.
Grâce à l’analyse des génomes mitochondriaux et nucléaires de fossiles vieux de 120 000 ans, nous retraçons les migrations, les contractions, les expansions, les extinctions et les remplacements des populations d’aurochs et de bisons. Nous cherchons à distinguer les effets de l’environnement et de l’humain sur l’évolution de ces grands herbivores en comparant deux espèces sœurs, Bos et Bison, car seul Bos (les bovins) a été domestique.
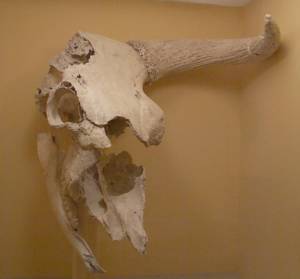
Nous avons reconstruit l’évolution phylogéographique du bison, montré l’influence des conditions environnementales sur la propagation et le remplacement des lignées ainsi que le comportement spécifique des mâles et femelles à l’origine de la discordance entre les lignées mitochondriales, les morphotypes et les génomes modernes, et identifié un ancêtre inconnu du bison européen actuel, qui a colonisé l’Europe après le dernier maximum glaciaire (LGM) il y a ~14 000 ans (Massilani et al., 2016, BMC Biology ; Grange et al., 2018, Diversity).
Nous avons caractérisé la diversité mitochondriale de l’aurochs sur un large échantillon de 125 individus en utilisant la capture de séquences de mitogénomes entiers, et de plus de 300 individus anciens par génotypage de mitotypes basé sur la PCR multiplexe. Cela nous a permis de suivre l’évolution des aurochs sauvages et des bovins domestiques dans le temps et l’espace en Europe, en Asie du Sud-Ouest et en Afrique du Nord, et de créer un cadre évolutif étendu en réponse aux changements climatiques des 50 000 dernières années et à l’interaction avec l’humain au cours des 10 000 dernières années (publication en prép.).
1.2. Paléogénomique des équidés.
Trois espèces d’équidés vivent encore dans l’hémisphère nord : les chevaux, les ânes sauvages d’Afrique et d’Asie. Alors que les chevaux et les ânes sauvages africains ont été domestiqués et sont omniprésents, les ânes sauvages asiatiques (hémiones) ne l’ont pas été et sont au bord de l’extinction. Nous avons identifié paléogénétiquement le moment de l’expansion des chevaux domestiques (Guimaraes et al., 2020) et l’histoire de la population des hémiones (Bennett et al., 2017). L’analyse paléogénomique des sépultures d’équidés de l’âge du Bronze en Mésopotamie et l’identification des populations d’hémiones éteintes nous ont permis de reconstituer les techniques d’élevage des premiers hybrides d’équidés connus à ce jour. Ces animaux étaient utilisés par l’élite dans les guerres entre les cités-états mésopotamiennes au 3ème millénaire avant notre ère (Bennett et al., 2022)
1.3. Paléogénomique de la domestication du chat.
En utilisant des génomes anciens, nous tentons de reconstruire l’histoire de la domestication du chat au cours des 10 000 dernières années, qui est très différente de celle des bovins. La domestication du chat a largement suivi la voie de la commensalité et l’élevage sélectif n’est devenu un mécanisme de domestication important que très récemment. En utilisant l’ADN mitochondrial ancien de spécimens archéologiques, nous avons reconstruit l’histoire de la propagation du chat sauvage d’Afrique du Nord et d’Asie du Sud-Ouest F.s.lybica, le prédécesseur du chat domestique : elle s’est déroulée en deux vagues, la première à partir du Croissant fertile au début du Néolithique, la seconde à partir de l’Égypte pendant l’Antiquité classique (Ottoni et al., 2017).
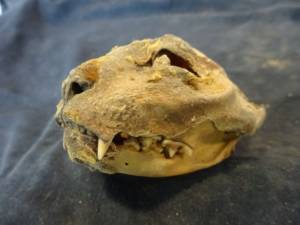
2.1. Morphologie des Denisoviens.
La découverte, grâce à la paléogénomique, d’une population humaine inconnue jusqu’alors, contemporaine des Néandertaliens, les Dénisoviens, a révolutionné la paléoanthropologie. Cependant, l’absence de restes squelettiques morphologiquement caractéristiques de cette population a éclipsé cette révolution. Nous avons analysé le mitogénome d’un os distal de doigt et l’avons identifié comme le spécimen type des Dénisoviens. C’était à l’époque le seul vestige squelettique qui pouvait être décrit morphologiquement. L’analyse réalisée par une paléoanthropologue collaboratrice a montré que les traits phénotypiques de l’os sont identiques à ceux de l’Homo sapiens et que les Néandertaliens ont développé des traits dérivés (Bennett et al., 2019).
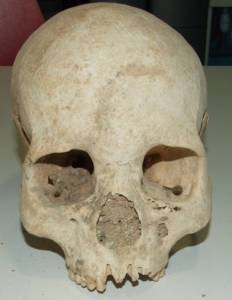
2.2. Paléogénomique du peuplement de l’Europe.
Nous avons analysé quatre périodes récentes importantes de transition culturelle et démographique dans l’histoire de l’humanité en Europe et exploré les traces enregistrées dans les génomes :
1) la période, il y a environ 40 000 ans, au cours de laquelle la première vague d’Homo sapiens ayant pénétré en Europe a connu en plus d’une période glaciaire un événement environnemental dramatique, l’éruption volcanique ignimbritique de Campanie (IC). Nous avons analysé les génomes de restes humains vieux de ~35 000 ans provenant de Crimée et caractérisé les affiliations génétiques avec les rares génomes disponibles à l’époque de l’éruption de l’IC pour proposer l’histoire du peuplement de l’Europe au cours du Paléolithique supérieur (Bennett et al., 2023).
2) La colonisation, il y a environ 7 000 ans, de l’Europe occidentale par les premiers agriculteurs néolithiques d’origine anatolienne qui se sont mélangés aux chasseurs-cueilleurs mésolithiques autochtones et ont introduit le mode de vie productif des sociétés agricoles qui est à la base de nos sociétés actuelles. Nous avons commencé cette ligne de recherche par une première analyse de génomes à faible couverture couvrant plusieurs régions du sud, de l’est et du nord de la France et datant de 9 000 à 2 000 ans (Brunel et al., 2020). Nous avons ensuite produit une centaine de génomes provenant de sites archéologiques néolithiques du Bassin parisien et reconstruit l’évolution des populations correspondantes, ainsi que les flux de gènes, les mélanges et les migrations qui ont eu lieu au cours de cette période. Cette étude nous permet de mieux comprendre l’évolution des populations et des sociétés correspondantes, car le Néolithique représente une période clé pour le développement des sociétés jusqu’à aujourd’hui. (Parasayan et al., en préparation)
3) L’introduction en Europe occidentale, il y a 4 500 ans, de l’ascendance des steppes européennes représente l’événement ultime de formation du génome européen. L’analyse génétique des populations des génomes d’une sépulture collective du Bassin parisien et la reconstruction du génome d’un ancêtre manquant dont l’ascendance steppique a été introduite dans une communauté néolithique nous ont permis de montrer la dynamique des mélanges entre les descendants des nomades des steppes et des agriculteurs néolithiques lors de cette transition culturelle majeure (Parasayan et al., 2024).
4) Enfin, nous avons produit les génomes d’une population médiévale de l’ouest de la France avant et après une période de famine, de guerre et d’épidémies au 14ème siècle. En particulier, nous avons analysé une population mérovingienne avec une incidence élevée de dysplasie de la hanche et avons caractérisé le profil génétique associée à ce syndrome à multiples facettes (Martin et al., in prep ; Parasayan et al., in prep).
2.3. Nous avons beaucoup investi dans le développement de méthodes appliquées à la recherche sur l’ADN ancien.
Nous avons inventé des adaptations clés des méthodes de construction de bibliothèques qui nous ont permis de construire très efficacement dans un format à haut débit plusieurs milliers de bibliothèques génomiques à partir d’échantillons très dégradés tout en évitant la plupart des pertes de matériel, inévitables lors de l’utilisation des méthodes actuelles. Nous avons appliqué certains de nos développements méthodologiques, dans le cadre d’une collaboration avec une équipe de recherche biomédicale, pour détecter l’ADN circulant dans le sang, une approche non invasive utilisée pour surveiller la progression des tumeurs. Cela a contribué à la découverte de mitochondries acellulaires circulant dans le sang humain (Al Amir Dache et al., 2020).
Nos projets de recherche s’inscrivent dans la continuité directe de nos réalisations et sont motivés par les questions relatives à l’évolution récente du génome des animaux domestiques et des humains depuis le Paléolithique supérieur, évoquées dans la section précédente. Notre nouvelle méthode de construction de bibliothèques adaptées aux traces d’ADN dégradé, qui sera publiée dans les années à venir, nous a permis d’obtenir plusieurs centaines de génomes anciens complets datant jusqu’à 120 000 ans. Nous allons maintenant nous concentrer sur l’étude de l’évolution de ces génomes, en particulier sur l’identification des régions génomiques sélectionnées au cours des 10 000 dernières années en utilisant des séries temporelles génomiques pour répondre aux questions suivantes :
1. Analyses génomiques de l’évolution de l’aurochs vers le bétail domestique.
L’aurochs, l’ancêtre sauvage des bovins domestiques, s’est éteint au XVIIe siècle en Europe centrale, et bien avant dans la plupart des régions inhabitées au début de l’Holocène. Sa domestication a débuté dans le sud-ouest de l’Asie, dans une région englobant la Turquie du sud-est, la Syrie du nord et l’Irak du nord actuels. Nous avons produit des génomes d’aurochs anciens à haute couverture, datés de 50 000 à 4 500 ans, et de bovins domestiqués entre 8 500 ans et le Moyen Âge, couvrant le sud-ouest, l’ouest et le sud-est de l’Europe, l’Anatolie, le Caucase et la vallée du Nil. Elles représentent les différentes étapes de la domestication, depuis les animaux sauvages d’origine jusqu’aux animaux domestiqués plus tardivement, et retracent ainsi l’évolution vers des races anciennes et modernes spécifiques. Afin de mieux documenter la diversité des races bovines actuelles qui n’ont pas fait l’objet d’une sélection intensive au cours des 200 dernières années, nous avons également produit cinq génomes de races rustiques modernes pour compléter les centaines de génomes de bovins modernes qui constituent notre ensemble de données, afin de suivre les changements génomiques qui se produisent en réponse à la domestication des bovins. Ces génomes sont maintenant placés dans le cadre de la population évolutive étendue que nous avons construit sur la base des mitogénomes. Nous analyserons en profondeur notre ensemble de données de séries temporelles génomiques afin d’identifier les régions sélectionnées aux différents stades du processus de domestication. Nous prendrons en compte la diversité des différentes populations d’aurochs qui ont pu participer à la formation des premiers génomes domestiques, à la fois en Asie du Sud-Ouest et pendant les migrations néolithiques des régions égéennes vers l’Europe de l’Ouest. Ces événements ont façonné le patrimoine génétique des humains et des animaux domestiques à l’époque préhistorique. Ces génomes anciens seront comparés à plus de 500 génomes de bovins domestiques modernes déjà disponibles et que nous avons traités parallèlement à nos génomes anciens. Nous utiliserons une grande diversité de méthodes de génomique des populations pour démêler la dérive des populations et les admixtures entre sauvages et domestiques. Nous analyserons aussi la sélection et identifierons ainsi les régions génomiques soumises à une pression sélective. Ce faisant, nous caractériserons dans le temps et dans l’espace la sélection exercée par différentes sociétés anciennes sur des régions génomiques particulières qui ont façonné la composition génomique des races bovines actuelles.
2. Analyses génomiques de l’évolution du bison européen.
Parallèlement à nos analyses de l’aurochs ancien, nous explorerons également le bison européen, une espèce étroitement apparentée, parce qu’elle n’a pas été domestiquée, qu’elle n’est pas encore éteinte et qu’elle nous permettra de contraster les trajectoires des changements évolutifs en réponse à la domestication ou à la pression exercée par l’humains sur l’habitat. À cette fin, nous avons généré des génomes de bisons européens anciens à couverture élevée, datant d’il y a 120 000 ans jusqu’au XVIIe siècle. Ils seront comparés à un ensemble de données comprenant des génomes de bisons européens actuels et des génomes de bisons américains. Cela nous permettra de suivre l’évolution génomique d’une population sauvage qui a subi de graves goulets d’étranglement en raison des changements climatiques et de la pression exercée par l’humain. Ce sera un contraste utile pour comprendre et interpréter correctement les changements génomiques de l’aurochs et des espèces bovines domestiquées.
3. L’évolution du génome du chat en réponse à la domestication.
Le deuxième modèle de domestication que nous étudierons en profondeur à l’échelle génomique est le chat. Une sélection intensive des caractères s’est produite chez cette espèce principalement depuis le 19e siècle seulement, même si certains traits comportementaux ont probablement été sélectionnés très tôt, mais pas nécessairement par l’intention de l’humain. Les événements génomiques sous-jacents associés à l’histoire de l’interaction des chats avec les humains seront caractérisés par l’analyse de génomes anciens générés à haute couverture et couvrant les 10 000 dernières années, ainsi que des génomes modernes de chats sauvages de France continentale et de Corse. Nous comparerons notre ensemble de données à un ensemble de génomes de chats modernes que nous avons traités en parallèle en utilisant des méthodes de génomique des populations similaires à celles du projet sur le génome du bétail.
4. Paléogénomique de l’évolution de la sensibilité et de la résistance aux maladies sur le territoire de la France actuelle depuis le Néolithique.
Nous étudierons l’évolution du génome humain au cours de la même période, en réponse aux changements de mode de vie qui ont suivi le passage des chasseurs-cueilleurs aux agriculteurs, c’est-à-dire la coévolution gène-culture associée aux changements de mode de vie (régime alimentaire riche en amidon et moins diversifié, famine, réponse immunitaire aux maladies infectieuses). L’objectif ultime est de comprendre comment l’évolution culturelle de l’humain a pu conduire l’évolution biologique humaine. En effet, ces changements de mode de vie ont affecté le régime alimentaire et l’exposition aux pathogènes, à la fois suite à l’augmentation de la densité des populations humaines, à l’augmentation des échanges à longue distance à travers le temps, et à l’interaction avec les animaux, en particulier en raison de la domestication, mais aussi en réponse à l’expansion des habitats humains empiétant sur ceux des animaux sauvages. Nous utiliserons à la fois les génomes humains néolithiques et médiévaux à couverture élevée de France que nous avons produits, mais nous inclurons également un certain nombre de génomes supplémentaires produits dans le domaine de la paléogénomique humaine. Le nombre de génomes humains disponibles est largement supérieur à celui des espèces domestiquées, et la puissance des méthodes de détection de la sélection est donc plus élevée. Ces données fournissent des informations sur le moment et la localisation de la sélection qui ne peuvent être obtenues par l’analyse des populations actuelles.
Membres
 Eva-Maria GEIGL, Emeritus researcher, GRANGE/GEIGL LAB+33 (0)1 57 27 81 32, bureau 583B
Eva-Maria GEIGL, Emeritus researcher, GRANGE/GEIGL LAB+33 (0)1 57 27 81 32, bureau 583B- Thierry GRANGE, Emeritus researcher, GRANGE/GEIGL LAB+33 (0)1 57 27 81 29, bureau 583B
Pour contacter un membre de l’équipe par mail : prenom.nom@ijm.fr
Parasayan, O., Laurelut, C., Bole, C., Bonnabel, L., Corona, A., Domenech-Jaulneau, C., Paresys, C., Richard, I., Grange, T.*, Geigl, E.-M.* (2024) Late Neolithic collective burial reveals admixture dynamics during the third millennium BCE and the shaping of the European genome Sci. Adv. 10, eadl2468 (2024). Doi: 10.1126/sciadv.adl2468
Bennett, E.A., Parasayan, O., Prat, S., Péan, S., Crépin, L., Yanevich, A., Grange, T.,*, Eva-Maria Geigl, E.-M.* Genome sequences of 36,000 to 37,000 year-old modern humans at Buran-Kaya III in Crimea. (2023) Nature Ecology and Evolution 7, 2160–2172. https://doi.org/10.1038/s41559-023-02211-9
Bennett, E.A., Weber, J., Bendhafer, W., Champlot, S., Peters, J., Schwartz, G., Grange, T., Geigl, E.-M. (2022) The genetic identity of the earliest human-made hybrid animals, the kungas of Syro-Mesopotamia. Science Advances 8, eabm0218. DOI : 10.1126/sciadv.abm0218
Guimaraes, S., Arbuckle, B., Peters, J., Adcock, S., Buitenhuis, H., Chavin, H., Manaseryan, N., Uerpmann, H.-P., Grange, T.*, Geigl, E.-M.* (2020) Ancient DNA shows domestic horses were introduced in the southern Caucasus and Anatolia during the Bronze Age. Science Advances, 6: eabb0030, DOI: 10.1126/sciadv.abb0030
Brunel, S., Bennett, E.A., Cardin, L., Garraud, D., Barrand Emam, H., Beylier, A., Boulestin, B., Chenal, F., Cieselski, E., Convertini, F., Dedet, B., Desenne, S., Dubouloz, J., Duday, H., Fabre, V., Gailledrat, E., Gandelin, M., Gleize, Y., Goepfert, S., Guilaine, J., Hachem, L., Ilett, M., Lambach, F., Mazière, F., Perrin, B., Plouin, S., Pinard, E., Praud, I., Richard, I., Riquier, V., Roure, R., Sendra, B., Thevenet, C., Thiol, S., Vauquelin, E., Vergnaud, L., Grange, T.*, Geigl, E.-M.*, Pruvost, M.* (2020) Ancient genomes from present-day France unveil 7,000 years of its demographic history. Proceedings of the National Academy of Sciences USA, 117(23):12791-12798, DOI:10.1073/pnas.1918034117.
Bennett, E.A., Crevecoeur, I., Viola, B., Derevianko, A.P., Shunkov, M.V., Grange, T., Maureille, B., Geigl, E.-M. (2019) Morphology of the Denisovan phalanx closer to modern humans than to Neandertals. Science Advances, 5:eaaw3950, DOI 10.1126/sciadv.aaw3950, DOI: 10.1126/sciadv.aaw3950
Grange, T., Brugal, J.-P., Flori, L., Gautier, M., Uzunidis, A., and Geigl, E.-M. (2018) The Evolution and Population Diversity of Bison in Pleistocene and Holocene Eurasia: Sex Matters. Diversity 10, 65; doi:10.3390/d10030065
Ottoni, C., Van Neer, W., De Cupere, B., Daligault, J., Guimaraes, S., Peters, J., Spassov, N., Prendergast, M.E., Boivin, N., Morales-Muniz, A., Bălăşescu, A., Becker, C., Benecke, N., Boronenanț, A., Buitenhuis, H., Chahoud, J., Crowther, A., Llorente, L., Manaseryan, N., Monchot, H., Onar, V., Osypińska, M., Putelat, O., Studer, J., Wierer, U., Decorte, R., Grange, T.*, Geigl, E.-M.* (2017) The paleogenetics of cat dispersal in the ancient world. Nature Ecology & Evolution 1, 0139. DOI: https://doi.org/10.1038/s41559-017-0139.
Bennett, E.A., Champlot, S., Peters, J., Arbuckle, B.S., Guimaraes, S., Pruvost, M., Bar-David, S., Davis, S.J.M., Gautier, M., Kaczensky, P., Kuehn, R., Mashkour, M., Morales-Muñiz, M., Pucher, E., Tournepiche, J.-F., Uerpmann, H.-P., Bălăşescu, A., Germonpré, M., Y. Günden, C., Hemami, M.-R., Moullé, P.-E., Öztan, A., Uerpmann, M., Walzer, C., Grange, T.*, Geigl, E.-M.* (2017) Taming the Late Quaternary phylogeography of the Eurasiatic wild ass through ancient and modern DNA. Plos one 12(4): e0174216. /doi.org/10.1371/journal.pone.0174216
Guimaraes S, Pruvost M, Daligault J, Stoetzel E, Bennett EA, Côté NM, Nicolas V, Lalis A, Denys C, Geigl E.-M.*, Grange T.* (2017) A cost-effective high-throughput metabarcoding approach powerful enough to genotype ~44 000 year-old rodent remains from Northern Africa. Mol Ecol Resour. 17: 405-417. doi: 10.1111/1755-0998.12565
Massilani, D., Guimaraes, S., Brugal, J.-P., Bennett, E.A., Tokarska, M., Arbogast, R., Baryshnikov, G., Boeskorov, G., Castel, J.-C., Davydov, S., Madelaine, S., Putelat, O., Spasskaya, N., Uerpmann, H.-P., Grange, T.*, Geigl,E.-M.* (2016) Past climate changes, population dynamics and the origin of Bison in Europe. BMC Biology 14:93-110. DOI 10.1186/s12915-016-0317-7
Côté, N.M.L., Daligault, J., Pruvost, M., Bennett, E.A., Gorgé, O., Guimaraes, S., Capelli, N., Le Bailly, M., Geigl, E.-M.*, Grange, T.* (2016) A New High-Througput Approach to Genotype Ancient Human Gastrointestinal Parasites. PLoS One. 2016 11(1):e0146230. doi: 10.1371/journal.pone.0146230. eCollection 2016.
Bennett, E.A., Massilani, D., Lizzo, G., Daligault, J., Geigl, E.-M., Grange, T. (2014) Library construction for ancient genomics: single strand or double strand? Biotechniques 56:289-300. DOI: 10.2144/000114176
Grange T. and Lourenço E. E. (2011) Mechanisms of Epigenetic Gene Activation in Disease: Dynamics of DNA Methylation and Demethylation, in Epigenetic Aspects of Chronic Diseases; pp 55-73; Roach H.I., Bronner F. and Oreffo R. O. eds, Springer, London.
Charruau P, Fernandes C, Orozco-Terwengel P, Peters J, Hunter L, Ziaie H, Jourabchian A, Jowkar H, Schaller G, Ostrowski S, Vercammen P, Grange T, Schlötterer C, Kotze A, Geigl, E.-M., Walzer C, Burger PA. (2010) Phylogeography, genetic structure and population divergence time of cheetahs in Africa and Asia: evidence for long-term geographic isolates. Mol Ecol. (4):706-724
Champlot S., Berthelot C., Pruvost M., Bennett E. A., Grange T. and Geigl E.M. (2010). An Efficient Multistrategy DNA Decontamination Procedure of PCR reagents for Hypersensitive PCR Applications. PLOS One, 5(9): e13042.
Mietton F, Sengupta AK, Molla A, Picchi G, Barral S, Heliot L, Grange T, Wutz A, Dimitrov S. (2009) Weak but uniform enrichment of the histone variant macroH2A1 along the inactive X chromosome. Mol Cell Biol. 29, 150-156
Grange T. (2008) Sensitive Detection of mRNA Decay Products Using Reverse-Ligation-Mediated PCR (RL-PCR). in Methods in Enzymology, RNA Turnover in Eukaryotes, Part B, 448, 445-466
Pruvost, M., Schwarz, R., Bessa Correia, V., Champlot, S., Grange, T. and Geigl, E.-M. (2008). DNA diagenesis and palaeogenetic analysis: critical assessment and methodological progress. Palaeogeography, Palaeoclimatology, Palaeoecology, 266, 211-219.
Miele V., C. Vaillant, Y. d’Aubenton-Carafa, C. Thermes and T. Grange (2008). DNA physical properties determine nucleosome occupancy from yeast to fly. Nucl Acids Res, 36, 3746-3756
Pruvost, M., T. Grange and E. M. Geigl (2005). « Minimizing DNA contamination by using UNG-coupled quantitative real-time PCR on degraded DNA samples: application to ancient DNA studies. » Biotechniques 38(4): 569-75.
abstract
Thomassin, H., C. Kress and T. Grange (2004). « MethylQuant: a sensitive method for quantifying methylation of specific cytosines within the genome. » Nucleic Acids Res 32(21).
abstract
Flavin, M., L. Cappabianca, C. Kress, H. Thomassin and T. Grange (2004). « Nature of the accessible chromatin at a glucocorticoid-responsive enhancer. » Mol Cell Biol 24(18): 7891-901.
abstract
Pruvost, M. and E. M. Geigl (2004). « Real-time quantitative PCR to assess the authenticity of ancient DNA amplification. » J. Archaeol. Sci. 31(9): 1191-1197.
abstract
Couttet, P. and T. Grange (2004). « Premature termination codons enhance mRNA decapping in human cells. » Nucleic Acids Res 32(2): 488-94. Print 2004.
abstract
Grange, T., L. Cappabianca, M. Flavin, H. Sassi and H. Thomassin (2001). « In vivo analysis of the model tyrosine aminotransferase gene reveals multiple sequential steps in glucocorticoid receptor action. » Oncogene 20(24): 3028-38.
abstract
Geigl, E.-M. (2002) « On the Circumstances Surrounding the Preservation and Analysis of Very Old DNA ». Archaeometry 44 (3) :337-342,
Thomassin, H., M. Flavin, M. L. Espinas and T. Grange (2001). « Glucocorticoid-induced DNA demethylation and gene memory during development. » Embo J 20(8): 1974-83.
abstract
Geigl, E. M. (2001). « Inadequate use of molecular hybridization to analyze DNA in Neanderthal fossils. » Am J Hum Genet 68(1): 287-91.
abstract
Christoffels, V. M., H. Sassi, J. M. Ruijter, A. F. Moorman, T. Grange and W. H. Lamers (1999). « A mechanistic model for the development and maintenance of portocentral gradients in gene expression in the liver. » Hepatology 29(4): 1180-92.
abstract
Sassi, H., R. Pictet and T. Grange (1998). « Glucocorticoids are insufficient for neonatal gene induction in the liver. » Proc Natl Acad Sci U S A 95(10): 5621-5.
abstract
Couttet, P., M. Fromont-Racine, D. Steel, R. Pictet and T. Grange (1997). « Messenger RNA deadenylylation precedes decapping in mammalian cells. » Proc Natl Acad Sci U S A 94(11): 5628-33.
abstract
De Sario, A., Geigl, E.-M., Palmieri, G., D’Urso, M., Bernardi, G.: A compositional map of human chromosome band Xq28. Proc. Natl. Acad. Sci. USA 93:1298-1302, 1996
Francke, U., Chang, E., Comeau, K., Geigl, E.-M., Giacalone, J., Li, X., Luna, J., Moon, A., Welch, S., Wilgenbus, P. (auteurs listés par ordre alphabétique) (1994) A radiation hybrid map of human chromosome 18. Cytogenet Cell Genet 66:196-213.
Leib-Mösch, C., Haltmeier, M., Werner, T., Geigl, E.-M., Brack-Werner, R., Francke, U., Erfle, V., Hehlmann, R. (1993) Genomic distribution and transcription of solitary HERV-K LTRs. Genomics 18:261-269.
Geigl, E.-M., Eckardt-Schupp, F. (1990) Chromosome-specific identification and quantification of S1 nuclease-sensitive sites in yeast chromatin by pulsed field gel electrophoresis. Molecular Microbiology 4(5):801-810.
Publications Thierry Grange
Kaptan D, Atağ G, Vural KB, Morell Miranda P, Akbaba A, Yüncü E, Buluktaev A, Abazari MF, Yorulmaz S, Kazancı DD, Küçükakdağ Doğu A, Çakan YG, Özbal R, Gerritsen F, De Cupere B, Duru R, Umurtak G, Arbuckle BS, Baird D, Çevik Ö, Bıçakçı E, Gündem CY, Pişkin E, Hachem L, Canpolat K, Fakhari Z, Ochir-Goryaeva M, Kukanova V, Valipour HR, Hoseinzadeh J, Küçük Baloğlu F, Götherström A, Hadjisterkotis E, Grange T, Geigl EM, Togan İZ, Günther T, Somel M, Özer F. The Population History of Domestic Sheep Revealed by Paleogenomes. Mol Biol Evol. 2024 Oct 4;41(10):msae158. doi: 10.1093/molbev/msae158. PMID: 39437846; PMCID: PMC11495565.
Latourte A, Jaulerry S, Combier A, Cherifi C, Jouan Y, Grange T, Daligault J, Ea HK, Cohen-Solal M, Hay E, Richette P. Ann Rheum Dis. (2024) SerpinA3N limits cartilage destruction in osteoarthritis by inhibiting macrophage-derived leucocyte elastase. ard-2024-225645. doi: 10.1136/ard-2024-225645. Online ahead of print. PMID: 39134394
Özkan M, Gürün K, Yüncü E, Vural KB, Atağ G, Akbaba A, Fidan FR, Sağlıcan E, Altınışık EN, Koptekin D, Pawłowska K, Hodder I, Adcock SE, Arbuckle BS, Steadman SR, McMahon G, Erdal YS, Bilgin CC, Togan İ, Geigl EM, Götherström A, Grange T, Özer F, Somel M. (2024) The first complete genome of the extinct European wild ass (Equus hemionus hydruntinus). Mol Ecol. 2024 Jul 1:e17440. doi: 10.1111/mec.17440.
Parasayan, O., Laurelut, C., Bole, C., Bonnabel, L., Corona, A., Domenech-Jaulneau, C., Paresys, C., Richard, I., Grange, T.*, Geigl, E.-M.* (2024) Late Neolithic collective burial reveals admixture dynamics during the third millennium BCE and the shaping of the European genome Sci. Adv. 10, eadl2468 (2024). Doi: 10.1126/sciadv.adl2468
Andrew Bennett, Oğuzhan Parasayan, Sandrine Prat, Stéphane Péan, Laurent Crépin, Alexandr Yanevich, Thierry Grange*, Eva-Maria Geigl* Genome sequences of 36,000 to 37,000 year-old modern humans at Buran-Kaya III in Crimea. (2023) Nature Ecology and Evolution 7, 2160–2172. https://doi.org/10.1038/s41559-023-02211-9
Jamieson A, Carmagnini A, Howard-McCombe J, Doherty S, Hirons A, Dimopoulos E, Lin AT, Allen R, Anderson-Whymark H, Barnett R, Batey C, Beglane F, Bowden W, Bratten J, De Cupere B, Drew E, Foley NM, Fowler T, Fox A, Geigl EM, Gotfredsen AB, Grange T, Griffiths D, Groß D, Haruda A, Hjermind J, Knapp Z, Lebrasseur O, Librado P, Lyons LA, Mainland I, McDonnell C, Muñoz-Fuentes V, Nowak C, O’Connor T, Peters J, Russo IM, Ryan H, Sheridan A, Sinding MS, Skoglund P, Swali P, Symmons R, Thomas G, Trolle Jensen TZ, Kitchener AC, Senn H, Lawson D, Driscoll C, Murphy WJ, Beaumont M, Ottoni C, Sykes N, Larson G, Frantz L. (2023) Limited historical admixture between European wildcats and domestic cats. Curr Biol. 33(21):4751-4760.e14. doi: 10.1016/j.cub.2023.08.031
Gorgé O., Bennett EA., Daligault J., Massilani D., Geigl E.-M.*, Grange T.* (2022) Analysis of ancient microbial DNA. Microbial Environmental Genomic, Methods Mol Biol 2605:103-131, doi: 10.1007/978-1-0716-2871-3_6
Bennett, E.A., Weber, J., Bendhafer, W., Champlot, S., Peters, J., Schwartz, G., Grange, T., Geigl, E.-M. (2022) The genetic identity of the earliest human-made hybrid animals, the kungas of Syro-Mesopotamia. Science Advances 8, eabm0218. DOI : 10.1126/sciadv.abm0218
Guimaraes, S., Arbuckle, B., Peters, J., Adcock, S., Buitenhuis, H., Chavin, H., Manaseryan, N., Uerpmann, H.-P., Grange, T.*, Geigl, E.-M.* (2020) Ancient DNA shows domestic horses were introduced in the southern Caucasus and Anatolia during the Bronze Age. Science Advances, 6: eabb0030, DOI: 10.1126/sciadv.abb0030
Al Amir Dache Z., Otandault A., Tanos R., Pastor B., Meddeb R., Sanchez C., Arena G., Lasorsa L., Bennett E.A., Grange T., El Messaoudi S., Mazard T., Prevostel C., Thierry A.R. (2020) Blood contains circulating cell-free respiratory competent mitochondria. FASEB J;34(3):3616-3630. doi: 10.1096/fj.201901917RR. Epub 2020 Jan 19.
Brunel, S., Bennett, E.A., Cardin, L., Garraud, D., Barrand Emam, H., Beylier, A., Boulestin, B., Chenal, F., Cieselski, E., Convertini, F., Dedet, B., Desenne, S., Dubouloz, J., Duday, H., Fabre, V., Gailledrat, E., Gandelin, M., Gleize, Y., Goepfert, S., Guilaine, J., Hachem, L., Ilett, M., Lambach, F., Mazière, F., Perrin, B., Plouin, S., Pinard, E., Praud, I., Richard, I., Riquier, V., Roure, R., Sendra, B., Thevenet, C., Thiol, S., Vauquelin, E., Vergnaud, L., Grange, T.*, Geigl, E.-M.*, Pruvost, M.* (2020) Ancient genomes from present-day France unveil 7,000 years of its demographic history. Proceedings of the National Academy of Sciences USA, 117(23):12791-12798, DOI:10.1073/pnas.1918034117.
Bennett, E.A., Crevecoeur, I., Viola, B., Derevianko, A.P., Shunkov, M.V., Grange, T., Maureille, B., Geigl, E.-M. (2019) Morphology of the Denisovan phalanx closer to modern humans than to Neandertals. Science Advances, 5:eaaw3950, DOI 10.1126/sciadv.aaw3950, DOI: 10.1126/sciadv.aaw3950
Geigl, E.-M., Grange, T. (2018) Ancient DNA: The quest for the best. Mol Ecol Resour. 18:1185–1187. DOI: 10.1111/1755-0998.12931
Grange, T., Brugal, J.-P., Flori, L., Gautier, M., Uzunidis, A., and Geigl, E.-M. (2018) The Evolution and Population Diversity of Bison in Pleistocene and Holocene Eurasia: Sex Matters. Diversity 10, 65; doi:10.3390/d10030065
Ottoni, C., Van Neer, W., De Cupere, B., Daligault, J., Guimaraes, S., Peters, J., Spassov, N., Prendergast, M.E., Boivin, N., Morales-Muniz, A., Bălăşescu, A., Becker, C., Benecke, N., Boronenanț, A., Buitenhuis, H., Chahoud, J., Crowther, A., Llorente, L., Manaseryan, N., Monchot, H., Onar, V., Osypińska, M., Putelat, O., Studer, J., Wierer, U., Decorte, R., Grange, T.*, Geigl, E.-M.* (2017) The paleogenetics of cat dispersal in the ancient world. Nature Ecology & Evolution 1, 0139. DOI: https://doi.org/10.1038/s41559-017-0139.
Bennett, E.A., Champlot, S., Peters, J., Arbuckle, B.S., Guimaraes, S., Pruvost, M., Bar-David, S., Davis, S.J.M., Gautier, M., Kaczensky, P., Kuehn, R., Mashkour, M., Morales-Muñiz, M., Pucher, E., Tournepiche, J.-F., Uerpmann, H.-P., Bălăşescu, A., Germonpré, M., Y. Günden, C., Hemami, M.-R., Moullé, P.-E., Öztan, A., Uerpmann, M., Walzer, C., Grange, T.*, Geigl, E.-M.* (2017) Taming the Late Quaternary phylogeography of the Eurasiatic wild ass through ancient and modern DNA. Plos one 12(4): e0174216. /doi.org/10.1371/journal.pone.0174216
Guimaraes S, Pruvost M, Daligault J, Stoetzel E, Bennett EA, Côté NM, Nicolas V, Lalis A, Denys C, Geigl EM*, Grange T.* (2017) A cost-effective high-throughput metabarcoding approach powerful enough to genotype ~44 000 year-old rodent remains from Northern Africa. Mol Ecol Resour. 17: 405-417. doi: 10.1111/1755-0998.12565
Massilani, D., Guimaraes, S., Brugal, J.-P., Bennett, E.A., Tokarska, M., Arbogast, R., Baryshnikov, G., Boeskorov, G., Castel, J.-C., Davydov, S., Madelaine, S., Putelat, O., Spasskaya, N., Uerpmann, H.-P., Grange, T.*, Geigl,E.-M.* (2016) Past climate changes, population dynamics and the origin of Bison in Europe. BMC Biology 14:93-110. DOI 10.1186/s12915-016-0317-7
Gorgé O, Bennett EA, Massilani D, Daligault J, Pruvost M, Geigl E.-M., Grange T. (2016) Analysis of Ancient DNA in Microbial Ecology. Methods Mol Biol. 1399:289-315. doi: 10.1007/978-1-4939-3369-3_17.
Côté, N.M.L., Daligault, J., Pruvost, M., Bennett, E.A., Gorgé, O., Guimaraes, S., Capelli, N., Le Bailly, M., Geigl*, E.-M., Grange, T.* (2016) A New High-Througput Approach to Genotype Ancient Human Gastrointestinal Parasites. PLoS One. 2016 11(1):e0146230. doi: 10.1371/journal.pone.0146230. eCollection 2016.
Guimaraes, S., Fernandez-Jalvo, Y., Stoetzel, E., Gorgé, O., Bennett, E.A., Denys, C., Grange, T., Geigl, E.-M. (2016) Owl pellets: a wise DNA source for small mammal genetics. Journal of Zoology 298(1):64–74. doi:10.1111/jzo.12285
Bennett, E.A., Massilani, D., Lizzo, G., Daligault, J., Geigl, E.-M., Grange, T. (2014) Library construction for ancient genomics: single strand or double strand? Biotechniques 56:289-300. DOI: 10.2144/000114176
Geigl E. M. and Grange T. (2012) Eurasian wild asses in time and space: Morphological versus genetic diversity. Ann Anat. 194:88-102
Geigl, E.M., Grange, T., Maureille, B. : Le génome néandertalien. Encyclopaedia Universalis, « La Science au présent 2011 », p. 133-140
Charruau P, Fernandes C, Orozco-Terwengel P, Peters J, Hunter L, Ziaie H, Jourabchian A, Jowkar H, Schaller G, Ostrowski S, Vercammen P, Grange T, Schlötterer C, Kotze A, Geigl EM, Walzer C, Burger PA. (2011) Phylogeography, genetic structure and population divergence time of cheetahs in Africa and Asia: evidence for long-term geographic isolates. Mol Ecol. (4):706-724
Grange T. and Lourenço E. (2011) Mechanisms of Epigenetic Gene Activation in Disease: Dynamics of DNA Methylation and Demethylation, in Epigenetic Aspects of Chronic Diseases; pp 55-73; Roach H.I., Bronner F. and Oreffo R. O. eds, Springer, London.
Mietton F, Sengupta AK, Molla A, Picchi G, Barral S, Heliot L, Grange T, Wutz A, Dimitrov S. (2009) Weak but uniform enrichment of the histone variant macroH2A1 along the inactive X chromosome. Mol Cell Biol. 29, 150-156
Champlot S., Berthelot C., Pruvost M., Bennett E. A., Grange T. and Geigl E.M. (2010). An Efficient Multistrategy DNA Decontamination Procedure of PCR reagents for Hypersensitive PCR Applications. PLOS One, 5(9): e13042.
Grange T. (2008) Sensitive Detection of mRNA Decay Products Using Reverse-Ligation-Mediated PCR (RL-PCR). In Methods in Enzymology, RNA Turnover in Eukaryotes, Part B, 448, 445-466
Pruvost, M., Schwarz, R., Bessa Correia, V., Champlot, S., Grange, T. and Geigl, E.-M.(2008) DNA diagenesis and palaeogenetic analysis: critical assessment and methodological progress. Palaeogeography, Palaeoclimatology, Palaeoecology, 266, 211-219.
Miele V., C. Vaillant, Y. d’Aubenton-Carafa, C. Thermes and T. Grange (2008) DNA physical properties determine nucleosome occupancy from yeast to fly. Nucl. Acids Res, 36, 3746-3756
Dugast-Darzacq C., and T. Grange (2008) MethylQuant : a real time PCR based method to quantify methylation at single specific cytosines. In Methods in Molecular Biology, DNA methylation: Methods and protocols, Second edition, J. Tost Ed, Humana Press, 507, 281-303.
Dugast-Darzacq C., T. Grange and N. B. Schreiber-Agus (2007) Differential effects of Mxi1-SR and Mxi1-SR in Myc antagonism. FEBS J. 274, 4643-53.
Pruvost, R. Schwarz, V. Bessa Correia, S. Champlot, S. Braguier, N. Morel, Y. Fernandez-Jalvo, T. Grange and EM Geigl (2007) Freshly excavated fossil bones are best for amplification of ancient DNA M. Proc. Ntl. Acad. Sci. USA, 104, 739-744
Hoogenkamp M, Gaemers IC, Schoneveld OJ, Das AT, Grange T, Lamers WH (2007) Hepatocyte-specific interplay of transcription factors at the far-upstream enhancer of the carbamoylphosphate synthetase gene upon glucocorticoid induction. FEBS J. 274, 37–45
Schalchi L., Grange T., Hänni, C., and Morange M. (2006) Bac to basics: l’ADN. La Recherche, 398, 75-78
Active cytosine demethylation triggered by a transcriptional regulator involves DNA strand breaks. Kress C., Thomassin H., and Grange T., (2006) Proc. Ntl. Acad. Sci. USA 103, 1112-1117.
Grange, T., J. Imbert and D. Thieffry (2005). « Epigenomics: large scale analysis of chromatin modifications and transcription factors/genome interactions. » Bioessays 27(11): 1203-5.
abstract
Flavin, M., L. Cappabianca, C. Kress, H. Thomassin and T. Grange (2004). « Nature of the accessible chromatin at a glucocorticoid-responsive enhancer. » Mol Cell Biol 24(18): 7891-901.
abstract
Thakur, N., V. K. Tiwari, H. Thomassin, R. R. Pandey, M. Kanduri, A. Gondor, T. Grange, R. Ohlsson and C. Kanduri (2004). « An antisense RNA regulates the bidirectional silencing property of the Kcnq1 imprinting control region. » Mol Cell Biol 24(18): 7855-62.
abstract
Thomassin, H., C. Kress and T. Grange (2004). « MethylQuant: a sensitive method for quantifying methylation of specific cytosines within the genome. » Nucleic Acids Res 32(21).
abstract
Pâques F. and T. Grange (2002). Architecture nucléaire et régulation transcriptionnelle chez les eucaryotes supérieurs. Med./Sci 18, 1245-1256.
abstract
Grange, T. (2001). Traduction nucléaire : un garde-fou contre les protéines tronquées ?
Med./Sci. 17, 914-915.
abstract
Grange, T., L. Cappabianca, M. Flavin, H. Sassi and H. Thomassin (2001). « In vivo analysis of the model tyrosine aminotransferase gene reveals multiple sequential steps in glucocorticoid receptor action. » Oncogene 20(24): 3028-38.
abstract
Kress, C., H. Thomassin and T. Grange (2001). « Local DNA demethylation in vertebrates: how could it be performed and targeted' » FEBS Lett 494(3): 135-40.
abstract
Thomassin, H., M. Flavin, M. L. Espinas and T. Grange (2001). « Glucocorticoid-induced DNA demethylation and gene memory during development. » Embo J 20(8): 1974-83.
abstract
Cappabianca, L., H. Thomassin, R. Pictet and T. Grange (1999). « Genomic footprinting using nucleases. » Methods Mol Biol 119: 427-42.
abstract
Christoffels, V. M., H. Sassi, J. M. Ruijter, A. F. Moorman, T. Grange and W. H. Lamers (1999). « A mechanistic model for the development and maintenance of portocentral gradients in gene expression in the liver. » Hepatology 29(4): 1180-92.
abstract
Grange T. and H. Thomassin (1999). « Comment déméthyler une cytosine? Enlève-t-on un peu, beaucoup, encore plus, ou pas du tout’ » Med./Sci., 15, 907-908.
abstract
Thomassin, H., E. J. Oakeley and T. Grange (1999). « Identification of 5-methylcytosine in complex genomes. » Methods 19(3): 465-75.
abstract
Christoffels, V. M., T. Grange, K. H. Kaestner, T. J. Cole, G. J. Darlington, C. M. Croniger and W. H. Lamers (1998). « Glucocorticoid receptor, C/EBP, HNF3, and protein kinase A coordinately activate the glucocorticoid response unit of the carbamoylphosphate synthetase I gene. » Mol Cell Biol 18(11): 6305-15.
abstract
Sassi, H., R. Pictet and T. Grange (1998). « Glucocorticoids are insufficient for neonatal gene induction in the liver. » Proc Natl Acad Sci U S A 95(10): 5621-5.
abstract
Bertrand, E., D. Castanotto, C. Zhou, C. Carbonnelle, N. S. Lee, P. Good, S. Chatterjee, T. Grange, R. Pictet, D. Kohn, D. Engelke and J. J. Rossi (1997). « The expression cassette determines the functional activity of ribozymes in mammalian cells by controlling their intracellular localization. » Rna 3(1): 75-88.
abstract
Bertrand, E., M. Fromont-Racine, R. Pictet and T. Grange (1997). « Detection of ribozyme cleavage products using reverse ligation-mediated PCR (RL-PCR). » Methods Mol Biol 74: 311-23.
abstract
Couttet, P., M. Fromont-Racine, D. Steel, R. Pictet and T. Grange (1997). « Messenger RNA deadenylylation precedes decapping in mammalian cells. » Proc Natl Acad Sci U S A 94(11): 5628-33.
abstract
Grange T., G. Rigaud, E. Bertrand, M. Fromont-Racine, M.-L. Espinás, J. Roux and R. Pictet (1997). “In vivo footprinting of the interaction of proteins with DNA and RNA.” in In vivo footprinting. Cartwright, I. L. Ed. Advances in Molecular and Cell Biology. JAI Press, Inc., Greenwich, Conn. 21, 73-109.
Grange, T., E. Bertrand, M. L. Espinas, M. Fromont-Racine, G. Rigaud, J. Roux and R. Pictet (1997). « In vivo footprinting of the interaction of proteins with DNA and RNA. » Methods 11(2): 151-63.
abstract
Espinas, M. L., J. Roux, R. Pictet and T. Grange (1995). « Glucocorticoids and protein kinase A coordinately modulate transcription factor recruitment at a glucocorticoid-responsive unit. » Mol Cell Biol 15(10): 5346-54.
abstract
Roux, J., R. Pictet and T. Grange (1995). « Hepatocyte nuclear factor 3 determines the amplitude of the glucocorticoid response of the rat tyrosine aminotransferase gene. » DNA Cell Biol 14(5): 385-96.
abstract
Sassi, H., M. Fromont-Racine, T. Grange and R. Pictet (1995). « Tissue specificity of a glucocorticoid-dependent enhancer in transgenic mice. » Proc Natl Acad Sci U S A 92(16): 7197-201.
abstract
Bertrand, E., M. Fromont-Racine, R. Pictet and T. Grange (1993). « Visualization of the interaction of a regulatory protein with RNA in vivo. » Proc Natl Acad Sci U S A 90(8): 3496-500.
abstract
Bertrand, E., R. Pictet and T. Grange (1994). « Can hammerhead ribozymes be efficient tools to inactivate gene function? » Nucleic Acids Res 22(3): 293-300.
abstract
Espinas, M. L., J. Roux, J. Ghysdael, R. Pictet and T. Grange (1994). « Participation of Ets transcription factors in the glucocorticoid response of the rat tyrosine aminotransferase gene. » Mol Cell Biol 14(6): 4116-25.
abstract
Schweizer-Groyer, G., A. Groyer, F. Cadepond, T. Grange, E. E. Baulieu and R. Pictet (1994). « Expression from the tyrosine aminotransferase promoter (nt -350 to 1) is liver-specific and dependent on the binding of both liver-enriched and ubiquitous trans-acting factors. » Nucleic Acids Res 22(9): 1583-92.
abstract
Fromont-Racine, M., E. Bertrand, R. Pictet and T. Grange (1993). « A highly sensitive method for mapping the 5′ termini of mRNAs. » Nucleic Acids Res 21(7): 1683-4.
abstract
Bertrand E., T. Grange and R. Pictet (1992). “Trans-acting hammerhead ribozymes in vivo: present limits and future directions.” in Gene regulation by antisense RNA and DNA. pp 71-81. R.P. Erickson & J. Izart Eds. Keistone Symposia series on Molecular and Cellular Biology. Raven Press, Ltd., New York.
Schweizer-Groyer, G., A. Groyer, F. Cadepond, T. Grange, E. E. Baulieu and R. Pictet (1992). « Two liver-enriched trans-acting factors support the tissue-specific basal transcription from the rat tyrosine aminotransferase promoter. » J Steroid Biochem Mol Biol 41(3-8): 747-52.
abstract
Grange, T., J. Roux, G. Rigaud and R. Pictet (1991). « Cell-type specific activity of two glucocorticoid responsive units of rat tyrosine aminotransferase gene is associated with multiple binding sites for C/EBP and a novel liver-specific nuclear factor. » Nucleic Acids Res 19(1): 131-9.
abstract
Rigaud, G., T. Grange and R. Pictet (1991). « Sequenase should be used instead of the Klenow fragment for the synthesis of oligonucleotides labeled to a high specific activity. » Nucleic Acids Res 19(17): 4777.
abstract
Rigaud, G., J. Roux, R. Pictet and T. Grange (1991). « In vivo footprinting of rat TAT gene: dynamic interplay between the glucocorticoid receptor and a liver-specific factor. » Cell 67(5): 977-86.
abstract
Grange, T., J. Roux, M. Fromont-Racine and R. Pictet (1989). « Positive and negative regulation of a transfected chimeric tyrosine aminotransferase gene: effect of copy number. » Exp Cell Res 180(1): 220-33.
abstract
Grange, T., J. Roux, G. Rigaud and R. Pictet (1989). « Two remote glucocorticoid responsive units interact cooperatively to promote glucocorticoid induction of rat tyrosine aminotransferase gene expression. » Nucleic Acids Res 17(21): 8695-709.
abstract
Oddos, J., T. Grange, K. D. Carr, B. Matthews, J. Roux, H. Richard-Foy and R. Pictet (1989). « Nucleotide sequence of 10 kilobases of rat tyrosine aminotransferase gene 5′ flanking region. » Nucleic Acids Res 17(21): 8877-8.
abstract
Grange, T., C. M. de Sa, J. Oddos and R. Pictet (1987). « Human mRNA polyadenylate binding protein: evolutionary conservation of a nucleic acid binding motif. » Nucleic Acids Res 15(12): 4771-87.
abstract
Rigaud, G., T. Grange and R. Pictet (1987). « The use of NaOH as transfer solution of DNA onto nylon membrane decreases the hybridization efficiency. » Nucleic Acids Res 15(2): 857.
abstract
Grange, T., M. Bouloy and M. Girard (1985). « Stable secondary structures at the 3′-end of the genome of yellow fever virus (17 D vaccine strain). » FEBS Lett 188(1): 159-63.
abstract
Grange, T., C. Guenet, J. B. Dietrich, S. Chasserot, M. Fromont, N. Befort, J. Jami, G. Beck and R. Pictet (1985). « Complete complementary DNA of rat tyrosine aminotransferase messenger RNA. Deduction of the primary structure of the enzyme. » J Mol Biol 184(2): 347-50.
abstract
Grange, T., F. Kunst, J. Thillet, B. Ribadeau-Dumas, S. Mousseron, A. Hung, J. Jami and R. Pictet (1984). « Expression of the mouse dihydrofolate reductase cDNA in B. subtilis: a system to select mutant cDNAs coding for methotrexate resistant enzymes. » Nucleic Acids Res 12(8): 3585-601.
abstract
Publications Eva-Maria Geigl
Leib-Mösch, C., Barton, D., Geigl, E.-M., Brack-Werner, R., Erfle, V., Hehlmann, R., Francke, U.: Two RFLPs associated with the human endogenous retroviral element S71 on chromosome 18q21. Nucl. Acid. Res 17 (6):2367, 1988
Hagen, U., Bertram, H., Geigl, E.-M., Kohfeldt, E., Wendel, S.: Radiation-induced clustered damage in DNA. Free Rad. Res. Comm. 6:177-178, 1989
Geigl, E.-M., Eckardt-Schupp, F. (1990) Chromosome-specific identification and quantification of S1 nuclease-sensitive sites in yeast chromatin by pulsed field gel electrophoresis. Molecular Microbiology 4(5):801-810.
Geigl, E.-M., Eckardt-Schupp, F. (1991) The repair of DNA double-strand breaks and S1 nuclease-sensitive sites can be monitored chromosome-specifically in Saccharomyces cerevisiae using pulsed-field gel electrophoresis. Molecular Microbiology, 5(7):1615-1620.
Geigl, E.-M., Eckardt-Schupp, F. (1991) Repair of gamma ray-induced S1 nuclease hypersensitive sites in yeast depends on homologous mitotic recombination and a RAD18-dependent function. Curr. Genet. 20:33-37.
Leib-Mösch, C., Haltmeier, M., Werner, T., Geigl, E.-M., Brack-Werner, R., Francke, U., Erfle, V., Hehlmann, R. (1993) Genomic distribution and transcription of solitary HERV-K LTRs. Genomics 18:261-269,
Francke, U., Chang, E., Comeau, K., Geigl, E.-M., Giacalone, J., Li, X., Luna, J., Moon, A., Welch, S., Wilgenbus, P. (auteurs listés par ordre alphabétique) (1994) A radiation hybrid map of human chromosome 18. Cytogenet Cell Genet 66:196-213,
De Sario, A., Geigl, E.-M., Bernardi, G. (1995) A rapid procedure for the compositional analysis of yeast artificial chromosomes. Nucleic Acids Res. 23(19):4013-4014,
De Sario, A., Geigl, E.-M., Palmieri, G., D’Urso, M., Bernardi, G. (J’ai co-encadré l’étudiante en these A. DeSario): A compositional map of human chromosome band Xq28. Proc. Natl. Acad. Sci. USA 93:1298-1302, 1996
Geigl, E.-M.: « Inadequate Use of Molecular Hybridization to Analyze DNA in Neanderthal Fossils ». Am. J. Hum. Genetics 68:287-290, 2001 ; S0002-9297(07)62499-9/10.1086/316948
Geigl, E.-M. : On the Circumstances Surrounding the Preservation and Analysis of Very Old DNA. Archaeometry 44 (3) :337-342, 2002
Geigl, E.-M., Baumer, U., and Koller, J.: New Appproaches to the study of the preservation of biopolymers in fossil bones. Environ. Chem. Letters 2(1) :45-48, 2004
Pruvost, M. and Geigl, E.-M. : Real-time Quantitative PCR to Assess the Authenticity of Ancient DNA, Journal of Archaeological Science, 31(9):1191-1197, 2004
Geigl, E.-M. and Pruvost, M. : Plea for a multidisciplinary approach to the study of Neolithic migrations : the analysis of biological witnesses and the input of palaeogenetics. In « Colonisation,, Migration, and Marginal Areas. A Zooarchaeological Approach. » M. Mondini, S. Munoz and S. Wickler (eds), Oxbow Books, p. 10-19, 2004.
Geigl, E.-M. : Why ancient DNA research needs taphonomy. In « Biosphere to Lithosphere: new studies in vertebrate taphonomy » T.O’Connor (ed.), Oxbow Books., p. 79-86, 2005.
Geigl, E.-M.: A personal analysis of the high failure rate of ancient DNA research. Geoarchaeological and Bioarchaeological Studies, Volume 3, p. 463-466, 2005.
Pruvost, M., Grange, T., and Geigl, E.-M.: Minimizing DNA-contamination by using UNG-coupled quantitative real-time PCR (UQPCR) on degraded DNA samples: application to ancient DNA studies. Biotechniques 38 :569-575, 2005; DOI 05384ST03
Pruvost, M., Schwarz, R., Bessa Correia, V., Champlot, S., Braguier, S., Morel, N., Fernandez-Jalvo, Y., Grange, T. and Geigl, E.-M.: Freshly excavated fossil bones are best for ancient DNA amplification. Proc. Natl. Acad. Sci. USA. 104 (3): 739-744, 2007 ; DOI 0610257104/ 10.1073/pnas.0610257104
Pruvost, M., Schwarz, R., Bessa Correia, V., Champlot, S., Grange, T. and Geigl, E.-M. DNA diagenesis and palaeogenetic analysis: critical assessment and methodological progress. Palaeogeography, Palaeoclimatology, Palaeoecology, 266 (3-4) : 211-219, 2008. doi:10.1016/j.palaeo.2008.03.041
Geigl, E.-M. : Palaeogenetics of cattle domestication : Methodological challenge for the study of fossil bones preserved in the domestication center in Southwest Asia. Comptes Rendus Palévol 7(2-3): 99-112, 2008
Fernandez-Jalvo, Y., Andrews, P., Pesquero, D., Smith, C., Marin-Monfort, D., Sanchez, B., Geigl, E.-M., Alonso, A. Early bone diagenesis in temperate environments. Part I: Surface features and histology. Palaeogeography, Palaeoclimatology, Palaeoecology 288:62-81, 2010
Champlot, S., Berthelot,C., Pruvost, M., Bennett, E.A., Grange, T., Geigl, E.-M. (2010) An Efficient Multistrategy DNA Decontamination Procedure of PCR reagents for Hypersensitive PCR Applications. PLoS ONE 5(9):e13042,. DOI 10.1371/journal.pone.0013042
Charruau, P., Fernandes, C., Orozco-Ter Wengel, P. Peters, J., Hunter, L., Ziaie, H., Jourabchian, A., Jowkar, H., Schaller, G., Ostrowski, S., Vercammen, P., Grange, T., Schlötterer, C., Kotze, A. Geigl, E.-M., Walzer, C., Burger, P.A. Phylogeography, genetic structure and population divergence time of cheetahs in Africa and Asia: evidence for long-term geographic isolation. Mol. Ecol. 20(4):706-24, 2010. doi: 10.1111/j.1365-294X.2010.04986
Geigl, E.-M. and Grange, T. (2012) Eurasian wild asses in time and space: morphological versus genetic diversity. Annals of Anatomy. 194:88-102,. doi:10.1016/j.aanat.2011.06.002
Bennett, E.A., Massilani, D., Lizzo, G., Daligault, J., Geigl, E.-M., Grange, T. (2014) Library construction for ancient genomics: single strand or double strand? Biotechniques 56:289-300. DOI: 10.2144/000114176
Nores, C., Morales-Muniz, A., Llorente Rodriguez, L., Bennett, E.A., Geigl, E.-M. (2015) The Iberian zebro: what kind of beast was it? Anthropozoologica 50(1):21-32.
Guimaraes, S., Fernandez-Jalvo, Y., Stoetzel, E., Gorgé, O., Bennett, E.A., Denys, C., Grange, T., Geigl, E.-M. (2016) Owl pellets: a wise DNA source for small mammal genetics. Journal of Zoology 298(1):64–74. doi:10.1111/jzo.12285
Côté, N.M.L., Daligault, J., Pruvost, M., Bennett, E.A., Gorgé, O., Guimaraes, S., Capelli, N., Le Bailly, M., Geigl*, E.-M., Grange, T.* (2016) A New High-Througput Approach to Genotype Ancient Human Gastrointestinal Parasites. PLoS One. 2016 11(1):e0146230. doi: 10.1371/journal.pone.0146230. eCollection 2016.
Gorgé O, Bennett EA, Massilani D, Daligault J, Pruvost M, Geigl EM, Grange T. (2016) Analysis of Ancient DNA in Microbial Ecology. Methods Mol Biol. 1399:289-315. doi: 10.1007/978-1-4939-3369-3_17.
Guimaraes S, Pruvost M, Daligault J, Stoetzel E, Bennett EA, Côté NM, Nicolas V, Lalis A, Denys C, Geigl EM*, Grange T.* (2017) A cost-effective high-throughput metabarcoding approach powerful enough to genotype ~44 000 year-old rodent remains from Northern Africa. Mol Ecol Resour. 17: 405-417. doi: 10.1111/1755-0998.12565
Massilani, D., Guimaraes, S., Brugal, J.-P., Bennett, E.A., Tokarska, M., Arbogast, R., Baryshnikov, G., Boeskorov, G., Castel, J.-C., Davydov, S., Madelaine, S., Putelat, O., Spasskaya, N., Uerpmann, H.-P., Grange, T.*, Geigl,E.-M.* (2016) Past climate changes, population dynamics and the origin of Bison in Europe. BMC Biology 14:93-110. DOI 10.1186/s12915-016-0317-7
Bennett, E.A., Champlot, S., Peters, J., Arbuckle, B.S., Guimaraes, S., Pruvost, M., Bar-David, S., Davis, S.J.M., Gautier, M., Kaczensky, P., Kuehn, R., Mashkour, M., Morales-Muñiz, M., Pucher, E., Tournepiche, J.-F., Uerpmann, H.-P., Bălăşescu, A., Germonpré, M., Y. Günden, C., Hemami, M.-R., Moullé, P.-E., Öztan, A., Uerpmann, M., Walzer, C., Grange, T.*, Geigl, E.-M.* (2017) Taming the Late Quaternary phylogeography of the Eurasiatic wild ass through ancient and modern DNA. Plos one 12(4): e0174216. /doi.org/10.1371/journal.pone.0174216
Ottoni, C., Van Neer, W., De Cupere, B., Daligault, J., Guimaraes, S., Peters, J., Spassov, N., Prendergast, M.E., Boivin, N., Morales-Muniz, A., Bălăşescu, A., Becker, C., Benecke, N., Boronenanț, A., Buitenhuis, H., Chahoud, J., Crowther, A., Llorente, L., Manaseryan, N., Monchot, H., Onar, V., Osypińska, M., Putelat, O., Studer, J., Wierer, U., Decorte, R., Grange, T.*, Geigl, E.-M.* (2017) The paleogenetics of cat dispersal in the ancient world. Nature Ecology & Evolution 1, 0139. DOI: https://doi.org/10.1038/s41559-017-0139.
Grange, T., Brugal, J.-P., Flori, L., Gautier, M., Uzunidis, A., and Geigl, E.-M. (2018) The Evolution and Population Diversity of Bison in Pleistocene and Holocene Eurasia: Sex Matters. Diversity 10, 65; doi:10.3390/d10030065
Geigl, E.-M., Grange, T. (2018) Ancient DNA: The quest for the best. Mol Ecol Resour. 18:1185–1187. DOI: 10.1111/1755-0998.12931
Bennett, E.A., Crevecoeur, I., Viola, B., Derevianko, A.P., Shunkov, M.V., Grange, T., Maureille, B., Geigl, E.-M. (2019) Morphology of the Denisovan phalanx closer to modern humans than to Neandertals. Science Advances, 5:eaaw3950, DOI 10.1126/sciadv.aaw3950, DOI: 10.1126/sciadv.aaw3950
Stoetzel, E., Lalis, A., Nicolas, V., Aulagnier,S., Benazzou, T., Dauphin, Y., Abdeljalil El Hajraoui, M., El Hassani, A., Fahd, S., Fekhaoui, M., Geigl, E.-M., Lapointe, F.-J., Leblois, R., Ohler, A., Nespoulet, R., Denys, C.. Quaternary terrestrial microvertebrates from mediterranean northwestern Africa: State-of-the-art focused on recent multidisciplinary studies (2019) Quaternary Science Reviews 224: 105966. DOI: 10.1016/j.quascirev.2019.105966
Brunel, S., Bennett, E.A., Cardin, L., Garraud, D., Barrand Emam, H., Beylier, A., Boulestin, B., Chenal, F., Cieselski, E., Convertini, F., Dedet, B., Desenne, S., Dubouloz, J., Duday, H., Fabre, V., Gailledrat, E., Gandelin, M., Gleize, Y., Goepfert, S., Guilaine, J., Hachem, L., Ilett, M., Lambach, F., Mazière, F., Perrin, B., Plouin, S., Pinard, E., Praud, I., Richard, I., Riquier, V., Roure, R., Sendra, B., Thevenet, C., Thiol, S., Vauquelin, E., Vergnaud, L., Grange, T.*, Geigl, E.-M.*, Pruvost, M.* (2020) Ancient genomes from present-day France unveil 7,000 years of its demographic history. Proceedings of the National Academy of Sciences USA, 117(23):12791-12798, DOI:10.1073/pnas.1918034117.
Guimaraes, S., Arbuckle, B., Peters, J., Adcock, S., Buitenhuis, H., Chavin, H., Manaseryan, N., Uerpmann, H.-P., Grange, T.*, Geigl, E.-M.* (2020) Ancient DNA shows domestic horses were introduced in the southern Caucasus and Anatolia during the Bronze Age. Science Advances, 6: eabb0030, DOI: 10.1126/sciadv.abb0030
Geigl, E.-M. (2021) PCR et paléogénétique : pour le meilleur et pour le pire. PCR and paleogenetics: for best and for worst. Bulletin de l’Académie Nationale de Médecine, 205 (4) :389-395.
Balasse, M., Bellot-Gurlet, L., Dillmann, Geigl, E.-M., Jacob, J., Lebon, M., Le Hô, A.-S., Lefèvre, J.-C., Leroyer, C., Mathé, V., Merle, V., Mirabaud, S., Reiche, I. (2021) Service, expertise et archéométrie : état d’une réflexion collégiale. ArchéoSciences 2, n° 45-2, 81 – 88.
Bennett, E.A., Weber, J., Bendhafer, W., Champlot, S., Peters, J., Schwartz, G., Grange, T., Geigl, E.-M. (2022) The genetic identity of the earliest human-made hybrid animals, the kungas of Syro-Mesopotamia. Science Advances 8, eabm0218. DOI : 10.1126/sciadv.abm0218
Gorgé O., Bennett EA., Daligault J., Massilani D., Geigl E.-M.*, Grange T.* (2022) Analysis of ancient microbial DNA. Microbial Environmental Genomic, Methods Mol Biol 2605:103-131, doi: 10.1007/978-1-0716-2871-3_6
Giorgi, C., Bayle, G. Gourichon, L., Ben Dhafer, W., Geigl, E.-M. (2023) Un assemblage de restes de bovins au sein de l’établissement élitaire laténien de Palaiseau « Les trois mares » (Essonne). Revue archéologique d’Île-de-France, 13 : 179-212.Bennett, E.A., Parasayan, O., Prat, S., Péan, S., Crépin, L., Yanevich, A., Grange, T.,*, Eva-Maria Geigl, E.-M.* Genome sequences of 36,000 to 37,000 year-old modern humans at Buran-Kaya III in Crimea. (2023) Nature Ecology and Evolution 7, 2160–2172. https://doi.org/10.1038/s41559-023-02211-9
Jamieson A, Carmagnini A, Howard-McCombe J, Doherty S, Hirons A, Dimopoulos E, Lin AT, Allen R, Anderson-Whymark H, Barnett R, Batey C, Beglane F, Bowden W, Bratten J, De Cupere B, Drew E, Foley NM, Fowler T, Fox A, Geigl EM, Gotfredsen AB, Grange T, Griffiths D, Groß D, Haruda A, Hjermind J, Knapp Z, Lebrasseur O, Librado P, Lyons LA, Mainland I, McDonnell C, Muñoz-Fuentes V, Nowak C, O’Connor T, Peters J, Russo IM, Ryan H, Sheridan A, Sinding MS, Skoglund P, Swali P, Symmons R, Thomas G, Trolle Jensen TZ, Kitchener AC, Senn H, Lawson D, Driscoll C, Murphy WJ, Beaumont M, Ottoni C, Sykes N, Larson G, Frantz L. (2023) Limited historical admixture between European wildcats and domestic cats. Curr Biol. 33(21):4751-4760.e14. doi: 10.1016/j.cub.2023.08.031
Parasayan, O., Laurelut, C., Bole, C., Bonnabel, L., Corona, A., Domenech-Jaulneau, C., Paresys, C., Richard, I., Grange, T.*, Geigl, E.-M.* (2024) Late Neolithic collective burial reveals admixture dynamics during the third millennium BCE and the shaping of the European genome Sci. Adv. 10, eadl2468 (2024). Doi: 10.1126/sciadv.adl2468
Özkan M, Gürün K, Yüncü E, Vural KB, Atağ G, Akbaba A, Fidan FR, Sağlıcan E, Altınışık EN, Koptekin D, Pawłowska K, Hodder I, Adcock SE, Arbuckle BS, Steadman SR, McMahon G, Erdal YS, Bilgin CC, Togan İ, Geigl EM, Götherström A, Grange T, Özer F, Somel M. (2024) The first complete genome of the extinct European wild ass (Equus hemionus hydruntinus). Mol Ecol. 2024 Jul 1:e17440. doi: 10.1111/mec.17440.
Kaptan D, Atağ G, Vural KB, Morell Miranda P, Akbaba A, Yüncü E, Buluktaev A, Abazari MF, Yorulmaz S, Kazancı DD, Küçükakdağ Doğu A, Çakan YG, Özbal R, Gerritsen F, De Cupere B, Duru R, Umurtak G, Arbuckle BS, Baird D, Çevik Ö, Bıçakçı E, Gündem CY, Pişkin E, Hachem L, Canpolat K, Fakhari Z, Ochir-Goryaeva M, Kukanova V, Valipour HR, Hoseinzadeh J, Küçük Baloğlu F, Götherström A, Hadjisterkotis E, Grange T, Geigl EM, Togan İZ, Günther T, Somel M, Özer F. The Population History of Domestic Sheep Revealed by Paleogenomes. Mol Biol Evol. 2024 Oct 4;41(10):msae158. doi: 10.1093/molbev/msae158. PMID: 39437846; PMCID: PMC11495565.
Book chapters
Eckardt-Schupp, F., Geigl, E.-M., Ahne, F., Siede, W.: Is mismatch repair involved in UV-inducible mutagenesis in yeast? in: DNA Replication and Mutagenesis. (Eds.: R.E. Moses, W.C. Summers). Washington: ASM, 355-361, 1988
Leib-Mösch, C., Bachmann, M., Geigl, E.-M., Brack-Werner, R., Werner, T., Erfle, V., Hehlmann, R.: Expression of S71-related sequences in human cells. Haematol. and Blood Transfusion Vol. 35, p. 256-259 in Modern Trends in Human Leukemia IX, R. Neth et al. (Eds.), Springer Verlag Berlin Heidelberg 1992
Geigl, E.-M. and Pruvost, M. : Plea for a multidisciplinary approach to the study of Neolithic migrations : the analysis of biological witnesses and the input of palaeogenetics. In « Colonisation,, Migration, and Marginal Areas. A Zooarchaeological Approach. » M. Mondini, S. Munoz and S. Wickler (eds), Oxbow Books, p. 10-19, 2004.
Geigl, E.-M. : Why ancient DNA research needs taphonomy. In « Biosphere to Lithosphere: new studies in vertebrate taphonomy » T.O’Connor (ed.), Oxbow Books., p. 79-86, 2005.
Geigl, E.-M.: The domestication of cattle: Insights from a joint archaeozoological-palaeogenetical venture. In “Between Sand and Sea. The Archaeology and Human Ecology of Southwestern Asia”. N.J. Conard, P. Drechsler and A. Morales (eds.), Kerns Verlag Tübingen, p. 69-90, 2011.
Geigl, E.-M. Lima de Guimaraes, S., Liesau, C. Palaeogenetic analysis of bovine remains from Camino de las Yeseras and Humanejos. In “Yacimientos Calcoliticos con campaniforme de la region de Madrid: nuevos estudios ». Blasco, C., Liesau, C., and Rios, P. (eds.). Universidad Autonoma de Madrid, Madrid, p. 199-210, 2011.
Geigl, E.-M., Champlot, S., de Lima Guimaraes, S., Bennett, E.A., Grange, T. (2013) Molecular Taphonomy of Spy: DNA Preservation in Bone Remains. In Rougier, H. & Semal, P., (eds.) Spy cave. 125 years of multidisciplinary research at the Betche aux Rotches (Jemeppe-sur-Sambre, Province of Namur, Belgium), Volume 1. Anthropologica et Præhistorica, 123/2012. Brussels, Royal Belgian Institute of Natural Sciences, Royal Belgian Society of Anthropology and Praehistory & NESPOS Society, pp. 371-380.
Bennett, E.A., Cardin, L., Geigl, E.-M. (2013) Etude paléogénétique des momies d’Antinoé conservées au Musée des Beaux Arts de Lille. In « Les momies d’Antinoé ». Lintz, Y., Coudert, M. (eds.), p. 99-102
Geigl, E.-M., and Grange, T. Taphonomie de l’ADN ancien. (2014) In « Manuel de Taphonomie ». C. Denys et M. Patou-Mathis (eds), Editions errance, Collection archéologiques, p. 147-164.
Geigl, E.M. (2015) La paléogénétique et paléogénomique. In « Messages d’os ». M. Balasse, J.-P. Brugal, Y. Dauphin, E.-M. Geigl, C. Oberlin, I. Reiche (eds), Editions des Archives contemporaines, p. 429-431
Geigl, E.M. and Grange, T. (2015) Les stratégies et enjeux de l’analyse de l’ADN ancien. In « Messages d’os ». M. Balasse, J.-P. Brugal, Y. Dauphin, E.-M. Geigl, C. Oberlin, I. Reiche (eds), Editions des Archives contemporaines, p. 433-452
Pruvost,M. and Geigl, E.-M. (2015) « Apport de la paléogénétique à l’étude des processus de domestication animale . In « Messages d’os ». M. Balasse, J.-P. Brugal, Y. Dauphin, E.-M. Geigl, C. Oberlin, I. Reiche (eds), Editions des Archives contemporaines, p. 469-485
Geigl, E.M. and Grange, T. (2015) Le génome des lignées humaines archaïques. In « Messages d’os ». M. Balasse, J.-P. Brugal, Y. Dauphin, E.-M. Geigl, C. Oberlin, I. Reiche (eds), Editions des Archives contemporaines, p. 501-520
Eva-Maria Geigl, Shirli Bar-David, Albano Beja-Pereira, E. Gus Cothran, Elena Giulotto, Halszka Hrabar, Tsendsuren Oyunsuren, Mélanie Pruvost (2016): Genetics of wild equids. In « Wild Equids: Ecology, Conservation, and Management », J. Ransom and P. Kazcensky (eds), p. 87-104
Geigl, E.-M., Guimaraes, S., Stoetzel, E., Fernandez-Jalvo, Y., Nespoulet, R., Denys, C., Grange, T. (2016) Analyse de la préservation de l’ADN dans les ossements des rongeurs de Témara, Maroc, en remontant dans le temps. In «Approche intégrative de la 6ème extinction : influence de l’installation des hommes modernes au Maroc sur l’évolution de la biodiversité des petits vertébrés terrestres ». Travaux de l’institut Scientifique de l’université Mohammed V de Rabat, Série générale, N° 8, 43-51
Geigl, E.-M., Bennett, E.A., Grange, T. (2016) Analyse de la degradation de l’ADN dans une phalange de la Dame du Cavillon. In “La grotte du Cavillon”. H. de Lumley (ed), CNRS Editions, p. 969-975.
Bennett, E. A., Gorgé, O., Grange, T., Fernández-Jalvo, Y. and Geigl, E.-M. (2016) Coprolites, Paleogenomics and Bone Content Analysis. In « Azokh Cave and the Transcaucasian Corridor». Yolanda Fernández-Jalvo, Tania King, Levon Yepiskoposyan, and Peter Andrews (eds), Springer, Vertebrate Paleobiology and Paleoanthropology Series, E. Delson and E.J. Sargis (eds), p. 271-286 DOI 10.1007/978-3-319-24924-7 (peer reviewed article)
Geigl, E.-M. & Grange, T. (2018) Of Cats and Men: Ancient DNA Reveals How the Cat Conquered the Ancient World. Charlotte Lindqvist and Om P. Rajora (eds.), Paleogenomics, Population Genomics [Om P. Rajora (Editor-in-Chief)], Springer International Publishing AG, part of Springer Nature 2018. DOI https://doi.org/10.1007/13836_2018_26 (peer reviewed article)
Geigl, E.-M. & Grange, T. (2019) Using palaeogenetics to unravel the impact of humans on animal populations in the past. In Animals: Cultural Identifiers in Ancient Societies? J. Peters, G. McGlynn & V. Goebel (eds)., Verlag Marie Leidorf (VML) GmbH, Rahden/Westf. p.131-137
Brunel, S., Bennett, E.A., Grange, T., Geigl, E.M., Pruvost, M. (2019) Etude paléogénomique des ossements du dolmen de Saint-Eugène. In Le dolmen de Saint-Eugène : autopsie d’une sépulture collective néolithique. Jean Guilaine., 1 vol., Archives d’écologie préhistorique, Toulouse, France. pp.339-345, 2020, 9782358420266 (pp. 405)
Liesau, C., Ríos, P., Vega, J., Blasco, C., Menduiña, R., de los Ángeles de Chorro, M., Cabrera, C., Geigl, E.-M., and Arteaga, C. (2022) The Bovine Deposits from the Chalcolithic Ditched Enclosure of Camino de las Yeseras (Madrid, Spain). In Wright, L. and Ginja, C. (eds) Cattle and People Interdisciplinary Approaches to an Ancient Relationship. Archaeobiology, 4, Series Editors Whitcher Kansa, S. and Lev-Tov, J., Lockwood Press, Columbus pp. 241-261.
Geigl, E.-M. (2023) Palaeogenetics and Palaeogenomics to Study the Domestication of Animals. In A. Mark Pollard, Ruth Ann Armitage, Cheryl A. Makarewicz (eds). Handbook of Archaeological Sciences DOI: 10.1002/9781119592112.ch33
PUBLICATIONS FOR THE GENERAL PUBLIC
1 Geigl, E.-M.: L’émergence de la paléogénétique. Biofutur 164:28-34, 1997
3 Geigl, E.-M., Grange, T., Maureille, B. : Le génome néandertalien. Encyclopaedia Universalis, « La Science au présent 2011 », p. 133-140
4 Geigl, E.-M. : Le métier du paléogénéticien. Archéothéma, Hors-série « Les métiers de l’archéologie » 2012, p. 58-59
5 « Mondes polaires – Hommes et biodiversités, des défis pour la science » (2012) R. Chenorkian, M. Raccurt (eds), Cherche-Midi
6 Geigl, E.-M. (2012) Les relations entre les humains et les animaux à la lumière de la génétique. L’archéologie au laboratoire. Coédition INRAP, Cité des Sciences et de l’Industrie Paris et éditions « La Découverte », p.61-74
7 Geigl, E.-M., Grange, T. (2013) Du potentiel mais des défis méthodologiques à relever. Biofutur 349, 24-27.
8 Geigl, E.-M. (2013) Conserver ou ressusciter les espèces en voie de disparition ? Biofutur 34, 32-34.
9 Pruvost, M., Massilani, D., Geigl, E.-M. (2013) La domestication retracée par l’ADN ancien. Biofutur 349, 36-39.
10 Geigl, E.-M. (2013) Quand étudier le génome des lignées humaines éteintes devient possible. Biofutur 349, 45-49.
11 Geigl, E.-M. (2015) L’apport de la paléogénétique et paléogénomique à l’archéologie. Nouvelles d’Archéologie 138, 10-14.
12 Geigl, E.-M., Bennett, E.A., Grange, T. (2015) Tracing the origin of our species through paleogenomics. BIO Web of Conferences 4, 00005 (2015) DOI: 10.1051/bioconf/20150400005. Owned by the authors, published by EDP Sciences, 2015
13 Geigl, E.-M. and Grange, T. (2016) La paléogénomique, pour une lecture du passé au présent. « L’empreinte du vivant » D. Joly, D. Faure, S. Salamitou (eds)., Editions Cherche Midi, p.151-165.
14 Geigl, E.-M. et Grange, T. (2017) Biodiversité passée et paléogénomique. « Les Big Data à Découvert », Bouzeghoub, M. et Mosseri, R. (eds), CNRS Editions, p. 228 – 229
15 Geigl, E.-M. et Grange, T. (2017) How cats conquered the Ancient world: a 9,000-years DNA tale. The Science Breaker. doi.org/10.25250/thescbr.brk062
16 Geigl, E.-M. (2017) Le peuplement de l’Europe vu par la paléogénomique. In « L’archéologie des migrations », Garcia D. and Le Bras, H. (eds.), La Découverte, Inrap, p. 67 – 80.
17 Geigl, E.-M. et Grange, T. (2017) « Comment le chat a conquis le monde ». Dans « De la Préhistoire à nos jours : Le Chat – Comment il a conquis le monde » Historia, Décembre 2017.
18 Grange, T. et Geigl, E.-M. (2018) « Grâce à la domestication, le chat a gagné le monde ». La Recherche, 531 :65-68
19 Geigl, E.-M. (2018) L’évolution des mammouths vue par la paléogénomique. In : Mémoire de Mammouth (244 p): [Exposition Musée national de Préhistoire – Les Eyzies de Tayac 29 juin – 12 novembre 2018] / Commissariat de l’exposition : Catherine Cretin, Stéphane Madelaine. Comité scientifique : Gennady Boeskorov, Peggy Bonnet-Jacquement, Jean-Jacques Cleyet-Merle, Philippe Fosse, Frédéric Plassard. Les Eyzies-de-Tayac, Musée national de Préhistoire. p. 31-34
20 Geigl, E.-M. (2018) La paléogénétique en tant qu’approche archéométrique au cours des 30 dernières années – Paleogenetics as Archeometrical approach over the last 30 years. ArcheoSciences, revue d’archéométrie, 42(1) : 95-104.
21 Grange, T. et Geigl, E.-M. (2019) « Grâce à la domestication, le chat a gagné le monde ». La Recherche – Les Essentiels, n° 30, p. 82-85
22 Geigl, E.-M. & Grange, T. (2019) Changements climatiques : 150 000 ans d’évolution des populations de bisons en Europe. In « 101 sécrets de l’ADN », Faure, D., Joly, D. Salamitou, S. (eds). Editions CNRS, p. 264-266.
23 Geigl, E.-M. (13/01/2020) « Une petite phalange réécrit l’histoire évolutive des humains », The Conversation https://theconversation.com/une-petite-phalange-reecrit-lhistoire-evolutive-des-humains-123156
24 Geigl, E.-M. (2020) « Le Peuplement de la France à la Protohistoire grâce à la paléogénomique ». Archéologia n° 589, 16-17.
25 Geigl, E.-M. (2020) “Qui a habité en France ces 9 000 dernières années ? » The Conversation https://theconversation.com/qui-a-habite-en-france-ces-9-000-dernieres-annees-139769
26 Geigl, E.-M. (2021) “Contribution de la paléogénétique à l’archéologie » In C. Carpentier, R.-M. Arbogast & Ph. Kuchler (dir.), Bioarchéologie : minimums méthodologiques, référentiels communs et nouvelles approches : actes du 4e séminaire scientifique et technique de l’Inrap, 28-29 nov. 2019, Sélestat. https://doi.org/10.34692/gdqj-7g88
27 Geigl, E.-M. (2023) “Crimea’s place in Europe 36,000 years ago: a tale of ancient genomes” https://ecoevocommunity.nature.com/manage/posts/211298
27 Geigl, E.-M., Grange, T. (2023) “Homo sapiens : comment deux crânes réécrivent l’histoire de son apparition en Europe – Nouvelle recherche » https://theconversation.com/homo-sapiens-comment-deux-cranes-reecrivent-lhistoire-de-son-apparition-en-europe-nouvelle-recherche-216195 ; https://theconversation.com/skulls-in-ukraine-reveal-early-modern-humans-came-from-the-east-216553
28 Geigl, E.-M., Parasayan, O., Grange, T. (2024) « Une tombe collective de 4 500 ans révèle son secret : la dernière étape de la formation du génome européen » https://theconversation.com/une-tombe-collective-de-4-500-ans-revele-son-secret-la-derniere-etape-de-la-formation-du-genome-europeen-232627
29 Geigl, E.-M., Parasayan, O., Grange, T. (2024) « A 4,500-year-old collective tomb in France reveals its secret – the final stage in the formation of the European genome » https://theconversation.com/a-4-500-year-old-collective-tomb-in-france-reveals-its-secret-the-final-stage-in-the-formation-of-the-european-genome-233382
30 Mattei, J., Grange, T., Geigl, E.-M. (2024) Le changement du rapport entre chat et humain au cours des 10000 dernières années. Revue semestrielle de droit animalier (RSDA) https://www.revue-rsda.fr/articles-rsda/7579-le-changement-du-rapport-entre-chat-et-humain-au-cours-des-10000-dernieres-annees
- Jeanne Mattei « Paléogénomique de la domestication des chats » soutenue le 20 décembre 2023
- Oguzhan Parasayan « Genomic evolution of of ancient French populations » soutenue le 20 décembre 2022
- Wejden Bendhafer : « Paléogénomique de l’évolution des Bovina et l’impact sur la domestication des bovins». Soutenance de thèse le 20/12/2021.
- Caitlin Martin : « The evolution of and selection upon the human genome since the invention of agriculture: a historical perspective on diseases”; en cours depuis Octobre 2020
- Samantha Brunel : « Analyse paléogénétique du peuplement de la France ». Soutenance de thèse le 14/11/2018.
- Diyendo Massilani : « Paléogénomique des Bovina et de leur domestication ». Soutenance de thèse le 6/7/ 2016.
- Olivier Gorgé : «Diagénèse de l’ADN bactérien et analyses métagénomiques de pathologies bactériennes du passé ». Soutenance de thèse le 13/12/2016.
- Nathalie Côté : « Apports de la paléogénétique à l’étude des helminthes garstro-interstinaux anciens». Soutenance de thèse le 16/12/2015 .
- Sophie Champlot : « Colonisation et domestication de l’Europe et du pourtour méditerranéen », Ecole Doctorale « Gènes, Génomes et Cellules », soutenue le 13/7/2010.
Collaborations scientifiques avec plus de 60 chercheurs nationaux et internationaux de 20 pays, surtout des archéologues, archéozoologues, paléoanthropologues.
Financements de l’équipe depuis 2010 :
2021-2024 Projet européen WIDESPREAD-05-2020 – Twinning: « Mapping The Neolithic Expansion In The Mediterranean: A Scientific Collective To Promote Archaeogenomics And Evolutionary Biology Research In Turkey” (H2020-WIDESPREAD-2020-5; CSA 952317; NEOMATRIX
2018-2021 “Paléogénomique de la domestication des bovins pour enrichir les stratégies durables de sélection (PATH2BOS) », ANR, coordinateur : Thierry Grange
2016-2021 “ Etude Génétique de la population Française (FROGH)“, ANR, coordinateur : Christian Dina, Institut du Thorax, UMR 1087/ UMR 6291, Nantes. Equipe « Epigénome & Paléogénome » : « Paléogénomique de la population médiévale de l’Ouest de la France ».
2015-2019 “Genetic characterization of Ancestral French populations using ancient DNA (ANCESTRA)” ANR JCJC – ANR-15-CE27-0001; Coordinatrice: Mélanie Pruvost, Institut Jacques Monod, Paris.
2014-2016 “The origins of Equid domestication”, National Science Foundation (NSF) Prime Award N° BCS-1311551; Coordinateur: Benjamin Arbuckle, University of Chapel Hill, North Carolina, USA. « Epigénome & Paléogénome » : “Paleogenetics of the domestication of horses in Anatolia and the Caucasus”
2010-2014 “Influence de l’installation des hommes modernes au Maroc sur l’évolution de la biodiversité des petits vertébrés terrestres (MOHMIE)“ ; Agence Nationale de Recherche (ANR) n° 2009 PEXT 004 05; Coordinatrice : C. Denys, Muséum National d’Histoire Naturelle. « Epigénome & Paléogénome » : “Paléogénétique des rongeurs au cours des derniers 120,000 ans”[/vc_column_text][/vc_tta_section][vc_tta_section title= »Actualités » tab_id= »1645025044634-e8b93c72-ce32″][vc_column_text]23 octobre 2023 – Nouvel article publié dans Nature Ecology and Evolution : Genome sequences of 36-37,000 year-old modern humans at Buran-Kaya III in Crimea
Vidéo sur la chaîne de Université Paris Cité : Paléogénomique : la double hélice comme machine à explorer le temps
19 novembre au 16 décembre 2024 : [Symphosium & Exposition ] Le peuplement de la France et les origines du mode de vie agricole
- Inscriptions gratuite mais obligatoire à eva-maria.geigl@ijm.fr
- exposition AGRIPOP
23 octobre 2023 – Nouvel article publié dans Nature Ecology and Evolution : Genome sequences of 36-37,000 year-old modern humans at Buran-Kaya III in Crimea
10 mars 2023 – Conférence au Collège de France » L’évolution des populations humaines et animales : une histoire de migrations et métissages » présenté dans le cadre du cours « L’histoire de l’humanité vue sous l’angle de la paléogénomique » donné par Lluis Quinatana-Murci.

A new study by the Grange/Geigl team published in Science Advances on 14 January 2022!
- Before the introduction of the domestic horse in Mesopotamia, valuable equids were being harnessed to ceremonial or military four wheeled wagons and used as royal gifts, but their true nature remained unknown.
- According to a palaeogenetic study, these prestigious animals were the result of a cross between a domestic donkey and a wild ass from Syria, now extinct.
- This makes them the oldest example of an animal hybrid produced by humans.
- Communiqué de presse
Fête de la science 2021 – Conférence « La génétique du peuplement de l’Europe depuis l’âge des glaces »
Communications de notre recherche par le CNRS :
https://archives.cnrs.fr/insb/article/2016/e-m-geigl
https://lejournal.cnrs.fr/articles/comment-le-chat-a-conquis-le-monde
https://www.cnrs.fr/fr/7-000-ans-dhistoire-demographique-en-france
https://www.cnrs.fr/fr/avant-les-chevaux-des-hybrides-danes-produits-pour-faire-la-guerre